
 2024-08-12
12:37:30
2024-08-12
12:37:30欧洲药品管理局(EMA)和互认与分散程序协调小组(CMDh)于5 月3日更新了“关于欧盟医疗器械法规Regulation (EU)2017/745和体外诊断器械法规Regulation(EU)2017/746实施情况的问答”, 本问答(Q&A)提供了药械组合产品在新的医疗器械法规(MDR)和体外诊断法规(IVDR)实施下不同情况的实际考虑,可以为药品、医疗器械和药械组合产品的申请人、上市许可人(MAH)和公告机构(NB)提供多方面的指导。
该指南指出,药械组合产品受医疗器械法规(EU)2017/745(MDR)或人用药品2001/83/EC指令的监管,具体的监管框架取决于该药械组合的主要作用模式。
· 对属于MDR第1(8)条第2段和第1(9)条第2段的整体组合产品(iDDC),若医疗器械作为iDDC的一个组成部分是单独使用的,同时药品的作用是主要的,则该iDDC将作为药品进行监管。比如与药品结合的可吞咽传感器。
如果产品的两个组成部分(医疗器械和药品)是以一个整体产品的方式投放市场,而且这个产品的组合方式是特定的并且其中的器械部分也不能重复使用,则该iDDC受药品法规监管。常见的此类产品有:预填充注射器、预填充笔、预填充特定药物的喷雾器等。
但是无论处于哪一法规框架的监管,iDDC产品的器械部分的安全和性能仍应符合MDR法规的GSPR要求。
· 有些情况下,药品和医疗器械在市场上被二次包装放置在一起,但不形成整体产品(iDDC),例如,一个含有药液的药瓶和一个带有CE标识的空的无菌注射器。则该无菌注射器需要按照医疗器械进行监管。
· 值得注意的是,体外诊断试剂盒不能包含药品。如果IVD产品或试剂盒打算与药品一起使用,即使这些产品是包装在一起的,IVD产品和药品也都必须各自符合其相应的法规。
· 边缘产品(borderline products)的分类由国家药品或医疗器械主管当局负责。
MDR法规自2021年5月26日正式实施,该法规第117条对药品2001/83/EC指令附录I第3.2章第12点进行了修订。该指南指出,MDR实施后,并不对在2021.5.26已授权的iDCC和2021.5.26前递交的iDDC申请产生影响,但它要求在2021年5月26日后递交的iDDC上市许可申请,以及上市后产生重大变更的iDDC(器械组件的设计、性能、安全性或预期用途可能对药品的交付或质量、安全性和功效产生重大影响,或者引入了新的器械)必须证明该器械部件符合下表所示的MDR法规的相关要求。
表1.涉及iDDC的上市许可申请变更摘要
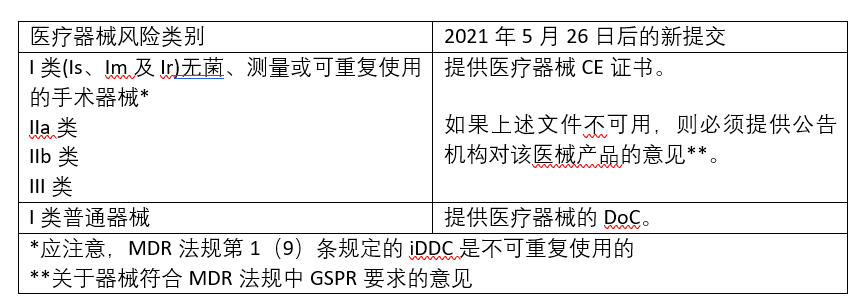
需要注意的是,药品委员会和国家主管部门不接受公告机确认器械产品部分符合GSPR的意见,必须是确认完全符合相关GSPR的意见;如果iDDC中包含了根据AIMDD指令或MDD指令颁发的CE证书的医疗器械,若该证书在MDR第120条的过渡条款下仍然有效,则可以提交以上表格中的文件以支持MDR第117条对2001/83/EC号指令进行的修订。
本指南还对iDDC上市后组件发生不同的变化后需要提交的文件和应注意的法规事项进行了详尽的举例说明。如想了解更详细的内容,您可以扫描以下二维码关注公众号进行进一步了解。

 / 地址:
/ 地址:
 / 电话:
+49251 322 66-64
/ 电话:
+49251 322 66-64  / 电邮:
contact@mednet-ecrep.com
/ 电邮:
contact@mednet-ecrep.com 













 EN
EN